Introduction
Bullous Pemphigoid (BP) is an autoimmune autoantibody-induced cutaneous inflammatory disease against BP180 or BP230 at the dermal-epidermal junction.1 Various Predisposing conditions like older age, poor general condition, dementia, high-dose corticosteroids and diabetes have been reported to be associated with this disease. 2 Clinical features include variably sized tense bullae of variable with clear or hemorrhagic fluid content, forming on erythematous or occasionally on healthy skin.3 These lesions are commonly observed in the lower abdomen, in the anterior and inner thighs and the flexor region of the forearm while Mucous membranes are affected in only a minority of the patients.4 A review of the literature has indicated that bullous pemphigoid patients, who are mostly treated with corticosteroids have at least one localized or systemic infection during the follow-up period including many fungal infections, however, the occurrence of mucormycosis is extremely rare and has a limited literature.5 We report a case which was clinically diagnosed as Bullous pemphigoid with a superimposed mucormycosis infection.
Case Presentation
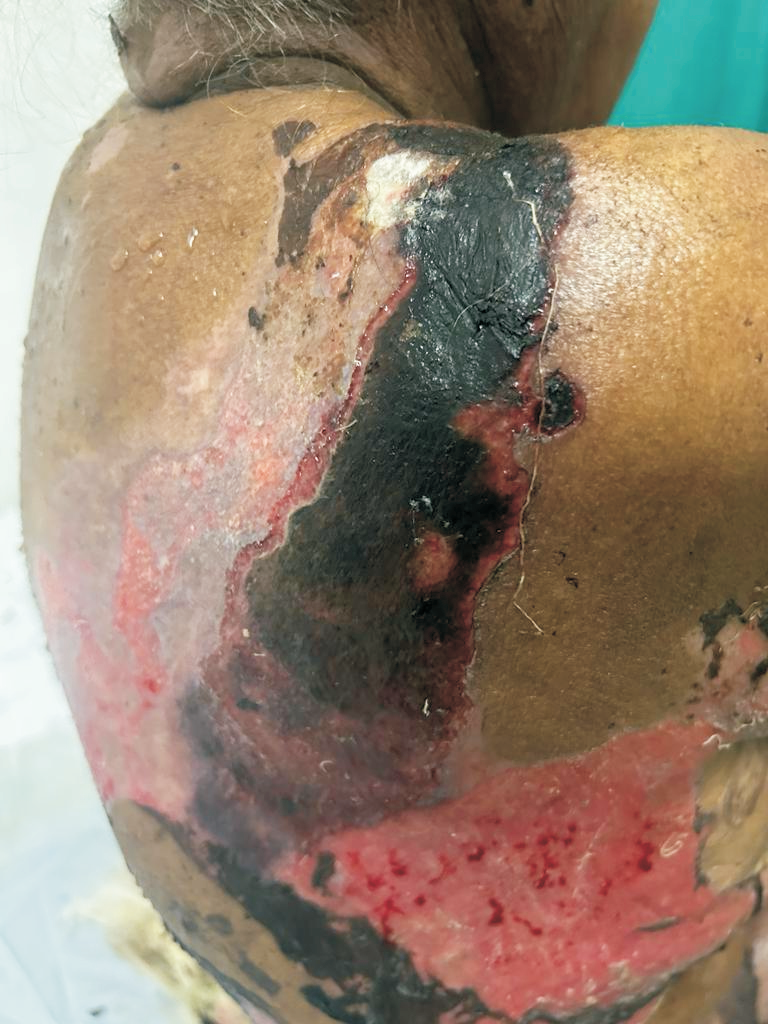
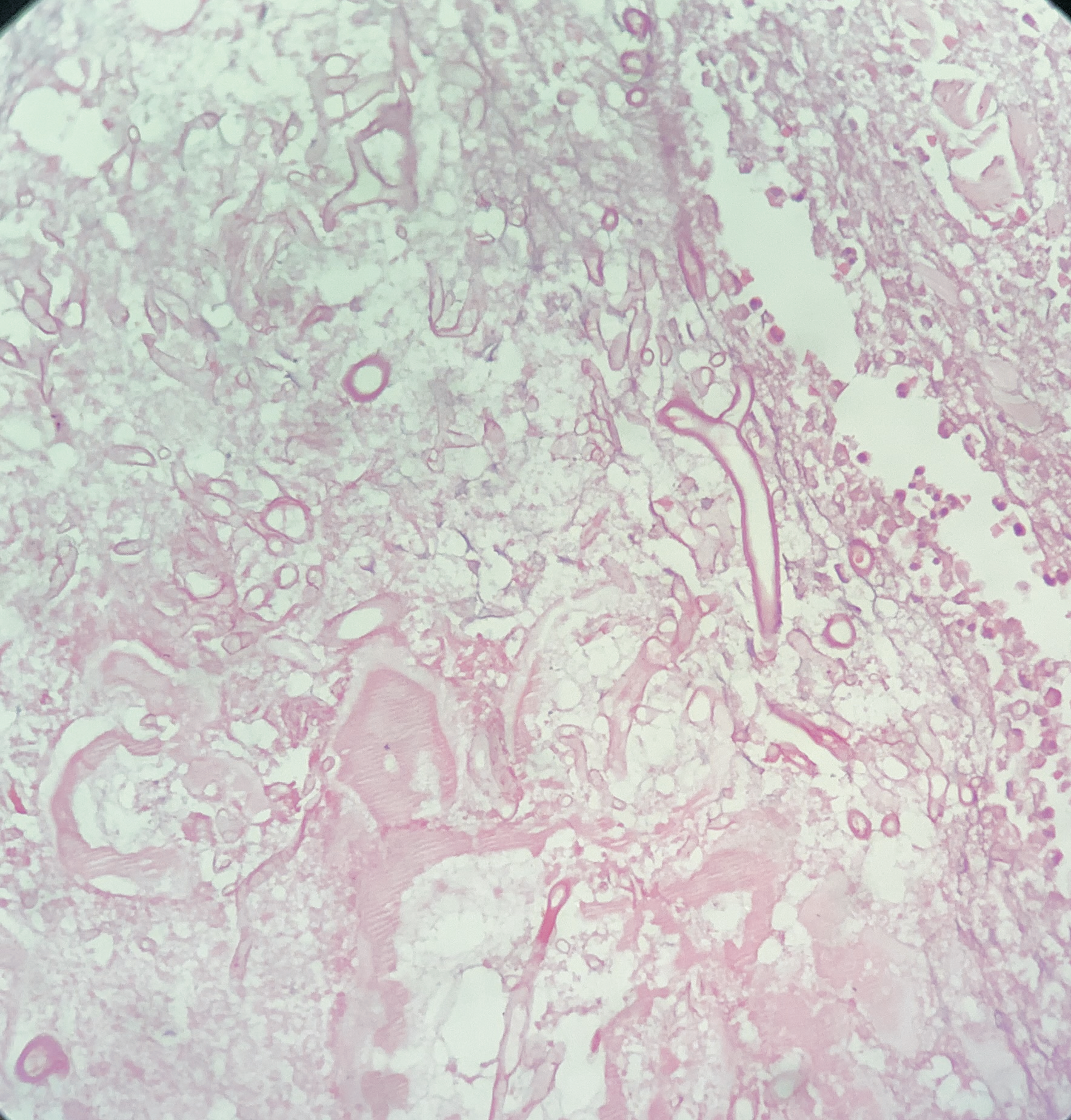
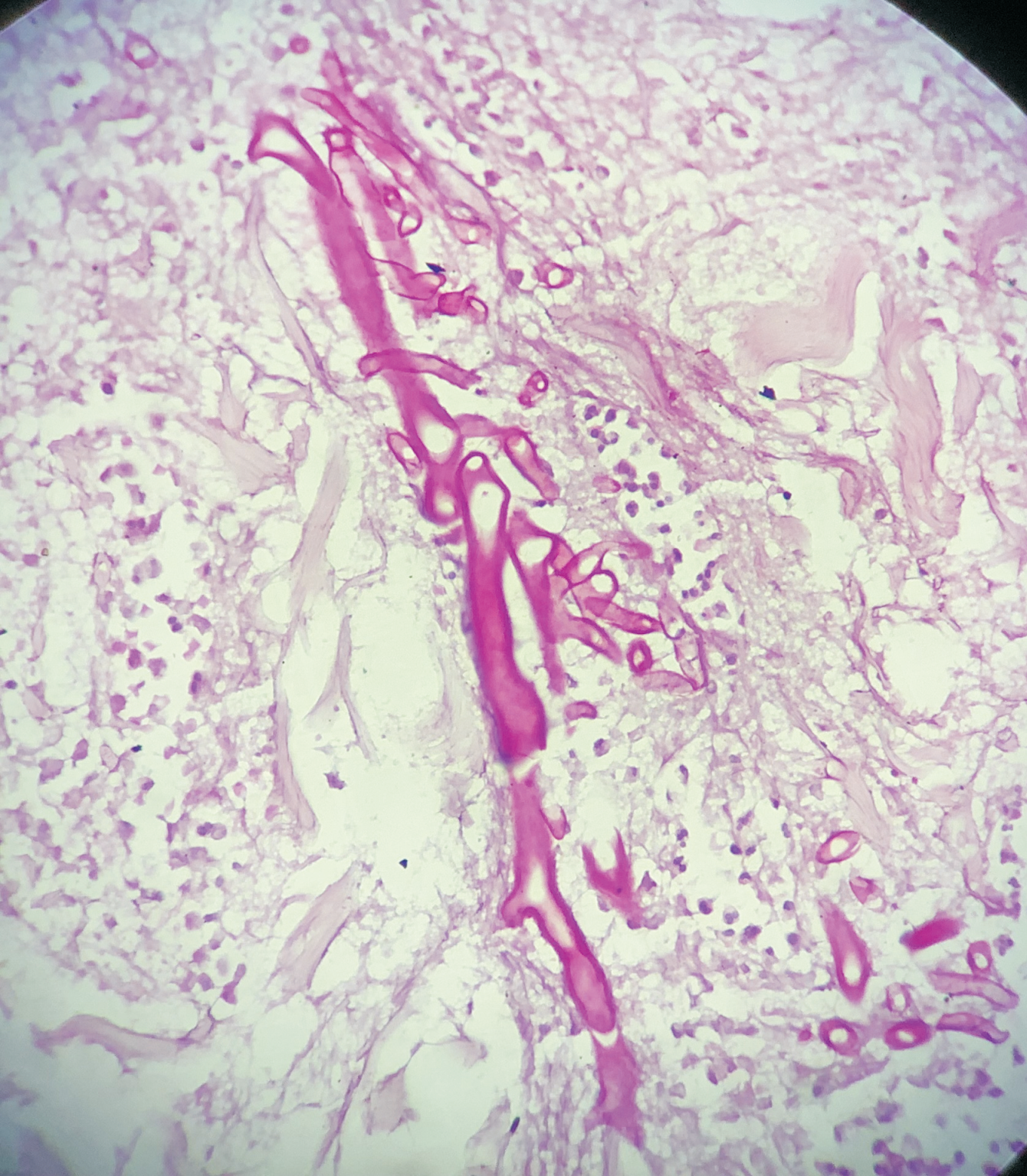
A 76-year-old female presented with multiple fluid-filled painful skin lesions all over the body for the past three months. The patient was normal for three months, later she developed multiple red raised itchy lesions over her hands followed by development on the chest. These lesions progressively increased in number and progressed to the whole body sparing the oral mucosa, genitals, palms, soles and scalp. This fluid-filled lesion did not rapture on pressure and tended to heal. The lesions were associated with pain, acute in onset, penetrating type with a moderate intensity which was relieved on taking medications. No history of pus discharge from these lesions neither there was any history of drug intake. She did not have any history of allergens. She was a known case of Diabetes Mellitus and hypertension. ECG was done which was suggestive of inferior wall ischemic changes for which cardiology reference was done after which 2D ECHO was done which was suggestive of CAD with concentric LVH with normal LVEF(56%) and mild MR. The patient was suggestive for CAG but she refused hence conservative management was done. On Examination Nikolsky's sign was negative while the bullae spread sign could not be elicited. A biopsy of the lesion was done which on histopathology revealed a subepidermal blister with eosinophilic-rich infiltrates in the papillary dermis. She was diagnosed with bullous pemphigoid and was treated with corticosteroids, antibiotics and topical ointments. On Post-treatment head to toe examination, there was a necrotic skin patch of about 4x4 cm over the right scapular region with a line of demarcation, with healthy surrounding skin with no active bleed or pus discharge (Figure 1). Surgery consultation was done and necrotic tissue was excised and sent for histopathological examination. Gross examination showed a fibro-fatty skin-covered tissue piece measuring 8.5 x 4.5 x 2.0 cm. The skin surface was necrosed. The cut surface was grey-brown to grey-yellow. When the microscopic examination was done sections showed ulcerated skin surface with necroinflammatory exudates viable epidermal lining or skin adnexa noted. Underneath the ulcerated surface, there was predominantly necrotic tissue, dense fibrin-inflammatory exudates, foci of fat necrosis, hhemorrhage, and congested to thrombosed blood vessels. Also noted are many scattered broad non-branched aseptate fungal hyphae infiltrating into fibro-fatty tissue collagenous septae and occasional vessels as well (Figure 2). PAS and GMS stain highlighted aseptate broad fungal hyphae confirming mucormycosis (Figure 3,4). Thus a final diagnosis of Invasive Mucormycosis with extensive necrosis was made. The patient was treated with anti-fungal drugs (Amphotericin B) and was kept on follow-up. After one month she succumbed to death due to coronary heart disease leading to heart failure.

Discussion
Bullous pemphigoid (BP) are rare autoimmune skin disease associated with high morbidity and mortality and is characterized by tense bullae of the skin and less oral involvement due to autoantibodies targeting hemi-desmosomal proteins (BP180 and BP230) anchoring the epidermis.6, 7 The etiopathogenesis of pemphigoid disease is complex and involves largely unknown environmental and polygenic genetic risk elements.8 HLA associations have been observed with BP, namely, DQB1*0301 has been highlighted in the literature multiple times as being associated with BP.9 The accurate epidemiologic and genetic risk factors for pemphigoid disease are still limited due to the rare and complex nature of these diseases, individual heterogeneity and variability of disease expression.10 BP presents with itchy, tense blisters over normal skin or erythematous and edematous background on the trunk and extremities affecting mainly the axillary folds, lower abdomen, inguinal areas and inner parts of the thighs, however, mucosal involvement is rare and is reported in 10-30% of the cases with oral, esophageal and genital lesions.11 Clinical evaluation of a patient with suspected BP involves a thorough dermatological examination of the skin and mucosa. Two disease scores are currently available for measuring BP activity, BP Disease Area Index (BPDAI) and Autoimmune Bullous Skin Disorder Intensity Score (ABSIS).12 Diagnosis relies on a careful correlation of clinical features suggestive of BP with immunopathological findings. Histopathology of non-bullous lesions usually demonstrates the presence of eosinophilic spongiosis with a mixed dermal inflammatory infiltrate; bullous lesions are usually characterized by subepidermal detachment with eosinophils, neutrophils and fibrin in the blister content, and dermal inflammatory infiltrate.13 Treatment of BP aims to arrest the development of new lesions and enable cutaneous healing and control of pruritus. As BP mainly affects elderly individuals, the choice of therapy has to be tailored according to the patient's comorbidities and ability to self-care to avoid potential complications and increased morbidity and mortality.14 Infections were found to be common and severe following the diagnosis of bullous pemphigoid. Patients with bullous pemphigoid were 3.3 times as likely to develop serious infections as those without bullous pemphigoid. Bullous pemphigoid per se doubled the risk of serious infections. Systemic corticosteroid use, the first-line therapy for bullous pemphigoid, was the strongest risk factor for serious infection in patients with bullous pemphigoid. Other independent risk factors associated with serious infections were advanced age, female sex, low income, and certain comorbidities. Prophylaxis of serious infections through active intervention with the risk factors might be essential in reducing the morbidity and mortality associated with bullous pemphigoid. 15