Introduction
Adrenal cortical carcinoma is a very rare malignant epithelial tumour of adrenal cortical cells. The reported annual incidence is only 0.5 - 2 cases per million population, however they account for upto 14% of adrenal incidentaloma.1 The median age of presentation is fifth to sixth decade with slight female predilection but cases have also been reported in children.2, 3 The clinical presentation can be varied and related to either hormone excess (cushiongoid features, hirsutism, virilization) or non specific symptomatology related to tumour growth (pain, abdominal fullness, general malaise).4
Most adrenal cortical carcinomas occur sporadically but rarely its association with hereditary syndromes like Li Fraumeni syndrome, Beckwith-Weidmann syndrome is reported in pediatric population, while in adults higher prevalence in association with Lynch syndrome, Multiple endocrine neoplasia type 1 (MEN 1) and Type 1 neurofibromatosis is well documented.5
Although radiological and histopathological characterization of adrenal cortical neoplasm as adenoma or carcinoma is quite straightforward, sometimes a benign tumour showing haemorrhage and revascularization on imaging and intermediate histology can pose a challenge. Histological diagnosis of adrenal cortical carcinoma is aided by a number of multiparametric scoring systems and diagnostic algorithms of which the Weiss system is most widely used.6 Some tumours commonly included in the differential diagnosis of adrenal cortical carcinoma include pheochromocytoma, renal cell carcinoma and sarcoma, hence immunohistochemistry plays an important role in the evaluation of adrenal masses.7
Case Summary
A 44 year old known hypertensive and hypothyroid female patient presented to the Gynaecology out patient department with complaints of bleeding per vaginum since 4 months and dull aching pain in the left lumbar region for the last 3 months. There was no history of fever, cough, vomiting, altered bowel movement or any urinary complaints. The past medical history was non significant. Per abdomen examination revealed mild tenderness in the left lumbar region while per vaginum and per speculum examination did not reveal any mass/ lesion.
Ultrasound showed a large heterogenous mass in the left upper quadrant of the abdomen. An abdominal CT scan showed a heterogenous lobulated mass, measuring 6.1 х 4.2 cm in the left suprarenal region inseparable from the upper pole of the left kidney and displacing it anteriorly. Further MRI showed a large, 10 х 6х 6 cm, well marginated, heterogenous left adrenal mass abutting the upper pole and posterior margin of left kidney (Figure 1). Due to patients' financial constraints, biochemical examinations to see the hormonal status of the tumour could not be performed. Based on the clinical and radiological findings, a provisional diagnosis of left sided pheochromocytoma with renal involvement was made and patient was taken up for left adrenalectomy.
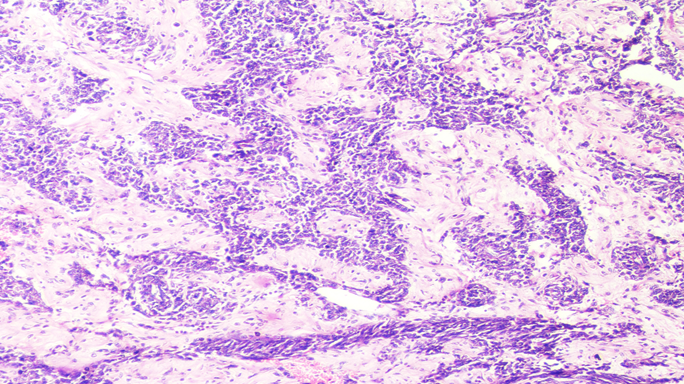
Grossly the specimen measured 12.5 х 8.2 х 3 cm in size with a well defined solid firm creamish white area of size 10.5 х 7.5 х 3 cm identified on cut section with foci of haemorrhage (Figure 2). Microscopic examination showed sheets and trabecular arrangement of tumour cells having mild to moderate anisonucleosis, hyperchromatic, irregular nuclei and moderate amount of eosinophilic cytoplasm (Figure 3). Prominent nucleoli was seen in some cells with <4 mitoses per 10 high power field. Diffuse foci of necrosis was present however no vascular invasion was noted. Sections also showed foci of extracellular myxoid background (Figure 4). Application of Weiss criteria for histological diagnosis gave a score of 3, which is consistent with malignant adrenal cortical tumour. Immunohistochemical expression of chromogranin showed negative staining (Figure 5) and pan cytokeratin showed faint focal cytoplasmic positivity (Figure 6). Based on histological morphology and immunohistochemistry, the case was signed out as myxoid variant of adrenocortical carcinoma.
Figure 1
MRI showed a large, 10 х 6х 6 cm, well marginated, heterogenous left adrenal mass abutting the upper pole and posterior margin of left kidney.
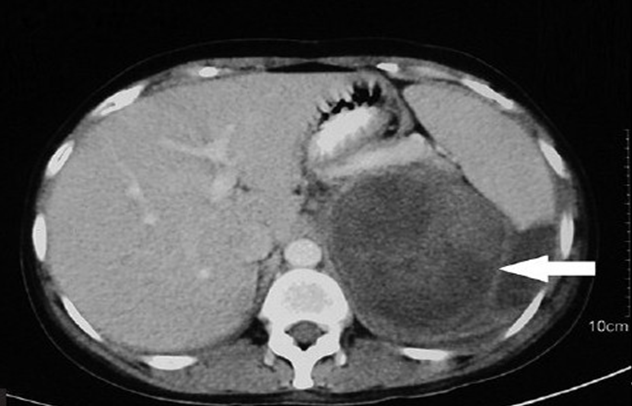
Figure 2
Grossly the specimen measured 12.5 х 8.2 х 3 cm in size with a well defined solid firm creamish white area of size 10.5 х 7.5 х 3 cm identified on cut section with foci of haemorrhage.
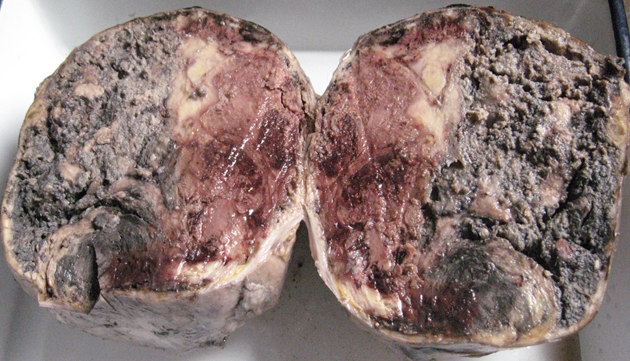
Figure 3
Microscopic examination showed sheets and trabecular arrangement of tumour cells having mild to moderate anisonucleosis, hyperchromatic, irregular nuclei and moderate amount of eosinophilic cytoplasm. Haematoxylin and Eosin x10X.



Discussion
We present a rare case of adrenocortical carcinoma in a middle aged female who presented with vaginal bleeding for 4 months. This abnormal vaginal bleeding could be attributed to the hormonal stimulation by the tumour however this rare tumour has been frequently reported to be non functional and the initial presentation is usually due to mass effects produced by the lesion.8 The common clinicalpresentations of adrenal cortical carcinoma include symptoms of hypercortisolism, hirsutism/ virilisation and general malaise.9 The initial presentation of the patient in this case prompted a gynaecological workup by the clinician.
Imaging plays a vital role in the assessment of adrenal masses. The adrenal cortical carcinomas usually measure >10 cm, are heterogenous and irregular masses with evidence of necrosis and haemorrhage within the tumour.10 The histopathological diagnosis of malignancy in adrenocortical tumours is aided by certain multifactorial diagnostic algorithms with high degree of concordance; Weiss score being the most popular one which takes into account parameters like nuclear grade, mitoses, percentage of clear cells, architecture, necrosis, vascular, sinusoidal and capsular invasion.11 Recently modified Weiss score proposed by Aubert et al. has also come into practice.12 The histological variants of adrenal cortical carcinoma include oncocytic, myxoid and sarcomatoid carcinoma, most common being oncocytic type. Myxoid adrenocortical carcinoma are very uncommon and often functional.13 Histologically this tumour is characterized by cords, clusters or pseudoglandular structures of tumour cells in a prominent myxoid background. Myxoid change in a tumour can vary from 10% to 95%.13
Immunophenotypically these tumours are similar to other variants, expressing markers specific for steroid producing cell like SF1, inhibin alpha, melan A and calretinin. Positivity for epithelial markers like cytokeratin, EMA and CEA is variable. Unlike chromogranin, synaptophysin is frequently expressed and hence its use for differentiation from pheochromocytoma is discouraged.14, 15 The tumours included in the differential diagnosis of adrenal cortical carcinoma include adrenal cortical adenoma and oncocytoma (which can be differentiated on histology alone), pheochromocytoma (chromogranin positive; negative in the case described), renal cell carcinoma (positive for epithelial markers; negative in the case described).
Some other tumours that can show myxoid change in this location include myxoma, liposarcoma, malignant fibrous histiocytoma and nerve sheath tumours etc. However careful histological, radiological and histological evaluation of the tumour alongwith an appropriate immunohistochemical panel including markers associated with adrenocortical differentiation can help to establish the correct diagnosis.15
Gene expression profiling of adrenocorticaltumours has elucidated the molecular pathogenesis of these tumours with emerging evidence for an adenoma to carcinoma progression. Studies have identified IGF2 as one of the most highly expressed genes in carcinoma while two other genes namely DLGAP5 and PINK1 have been identified in adrenocortical tumours of uncertain malignant potential and related with recurrence.16, 17
Surgery is the mainstay of treatment and the only potentially curative option. Adjuvant adrenolytic treatment with mitotane is offered to patients with incomplete/uncertain resection which is shown to reduce the risk of local recurrence.10 Radiotherapy of tumour bed is recommendedin incompletely resected tumours or those with complete resection but high risk (microscopic evidence of vascular invasion or Ki-67 >10%).18 The five year overall survival of patients with adrenocortical carcinoma is below 40%. The European Network for the Study of Adrenal Tumours (ENSAT) Staging Score, which assesses the extent of disease at the time of diagnosis is utilized as a major determinant of survival.Other prognostic factors include tumour grade, mitotic count and ki-67 proliferation index.19 p53 and CTNBB1 immunohistochemistry has been widely utilized as surrogates for molecular alteration in the two major signalling pathways of adrenocortical carcinoma, which also correlate with aggressive phenotypes and poor survival. 20
Conclusions
Adrenocortical carcinomas are rare tumours with varied clinical presentations or discovered incidentally. Tumours with unusual histology like myxoid need to be identified because of its rarity and to initiate immediate therapeutic interventions. Proper radiological and histological evaluation together with application of a panel of markers is indispensable in the work up of these adrenal masses.